六亚甲基二异氰酸酯(HDI)三聚体是高性能聚氨酯固化剂的核心组分,广泛应用于高档汽车涂装、航空复合材料、高端家具及体育器材等领域。与芳香族异氰酸酯固化剂相比,HDI三聚体具有低挥发、低毒性、优异耐候性和热稳定性等突出优势。然而,HDI自聚反应过程复杂,易产生二聚体、不对称三聚体、五聚体、七聚体等副产物,严重影响产品粘度与固化性能。因此,深入揭示其本征反应机理及催化剂作用规律,是实现高效可控生产的关键。
青岛科技大学毕荣山教授、徐盼副教授团队采用实验表征与分子模拟相结合的策略,系统开展了从“本征机理”到“催化强化”的完整理论研究。基于Gaussian软件进行密度泛函理论(DFT)计算,在B3LYP/6-31G(d)基组水平上系统模拟了HDI三聚体及多种副产物的生成路径,证明了HDI三聚反应中两步路径为最优反应路径。在催化机理研究过程中,团队运用多种分子模拟手段,从静电相互作用、弱相互作用及电子结构三个分子层次,深入揭示了乙酸钾与苄基三甲基氢氧化铵两类催化剂在HDI三聚反应中的微观作用机制。通过DFT计算、过渡态搜索、IRC路径验证及KiSThelP速率常数计算,明确了苄基三甲基氢氧化铵催化剂在关键能垒和反应速率常数上的显著优势,为高选择性HDI三聚催化剂的理性设计与工艺优化提供了理论依据。
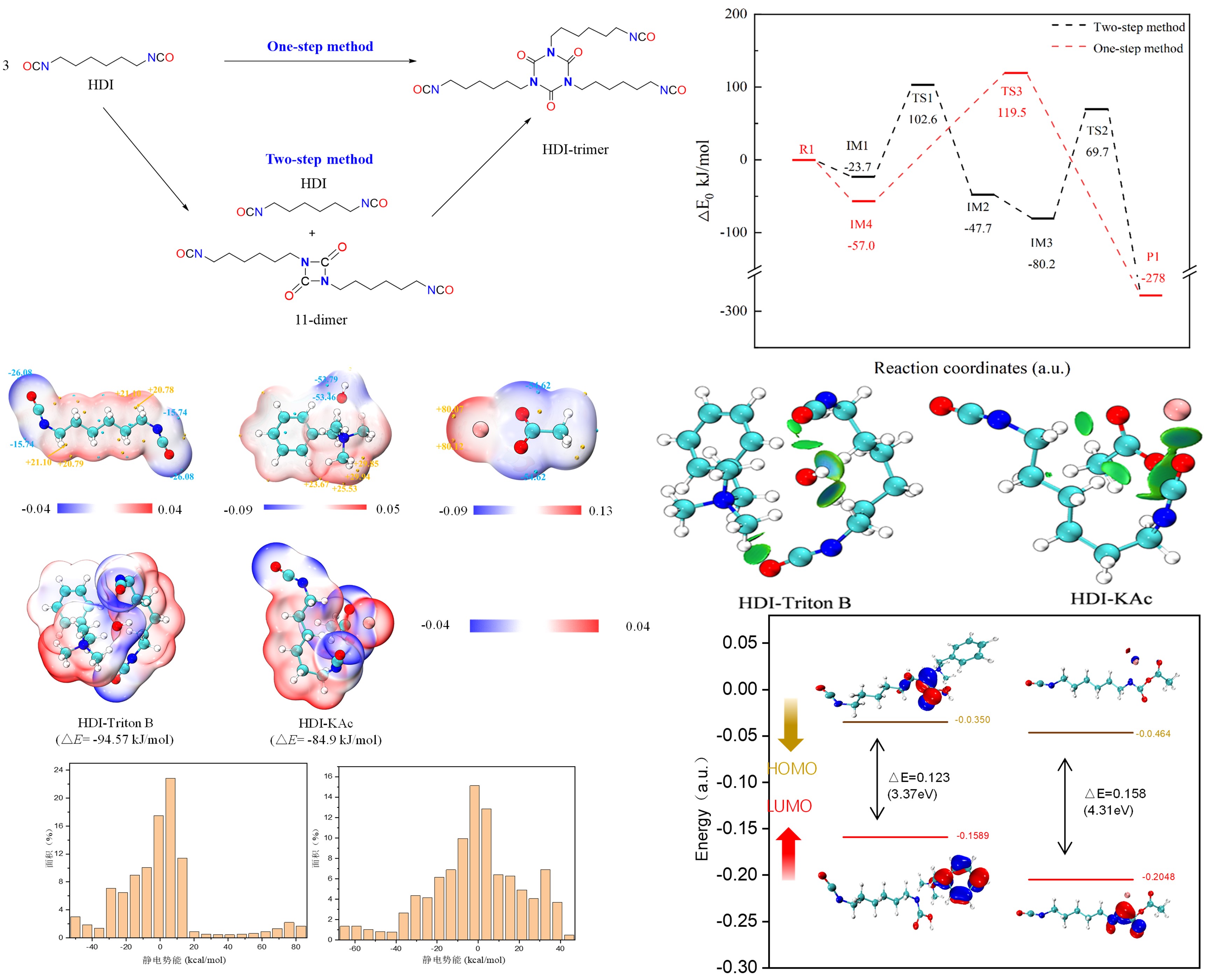
相关成果分别发表于化工三大核心期刊《Industrial & Engineering Chemistry Research》和《Chemical Engineering Science》上,青岛科技大学2023级硕士研究生石冉冉为两篇论文的第一作者,毕荣山教授、徐盼副教授为共同通讯作者。
原文链接如下:https://doi.org/10.1021/acs.iecr.5c01301
https://doi.org/10.1016/j.ces.2026.124338
 化工学院微信
化工学院微信