糖尿病是全球高发的代谢性疾病,开发长效、高效、无副作用的抗糖尿病药物是当前重大需求。传统多肽药物面临半衰期短、细胞膜穿透性差、溶解度低等瓶颈,尤其针对细胞内靶点(如蛋白酪氨酸磷酸酶PTPN1/2)的药物设计更是挑战。蛋白酪氨酸磷酸酶PTPN1/2是调控胰岛素和瘦素信号通路的关键细胞内靶点,但其高度保守的带负电活性位点使得传统小分子药物难以有效靶向,被认为是“不可成药”靶点。肽类药物在代谢疾病治疗中表现出巨大潜力,但面临膜通透性差、半衰期短和易被酶降解等问题。尽管脂肪酸修饰(如长链脂肪酸)可延长肽类药物的半衰期(如GLP-1受体激动剂司美格鲁肽),但其对细胞内靶点的通透性提升有限。因此,开发兼具长效性和细胞通透性的肽类抑制剂仍是当前研究的难点。
针对上述难点问题,在前期工作的基础上(J. Med. Chem.2023, 66, 4, 3030–3044.;ACS Pharmacol. Transl. Sci.2024, 7 (5), 1426-1437.),化工学院张传亮副教授与生物工程学院顾玉超教授团队创新性地提出双脂肪酸偶联和脂肪酸糖基化双偶联等协同修饰策略,成功设计、开发靶向细胞内PTPN1/2的超长效多肽降糖药物,在靶向“不可成药”靶点的超长效降糖药物方向取得新进展。
进展一:提出了一种“双脂肪酸共轭”策略,通过将长链脂肪酸(C16)与中链脂肪酸(C10-C12)分别修饰在BimBH3肽的N端和Lys²侧链上,设计了一系列新型肽类抑制剂。长链脂肪酸增强代谢稳定性和长效性,中链脂肪酸提升细胞通透性和靶向性,克服了单一修饰的局限性。
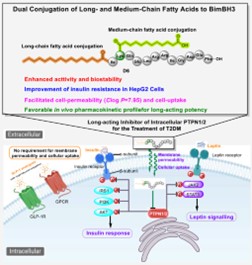

优化后的化合物D6对PTPN1/PTPN2表现出双重抑制活性,并通过分子对接揭示了其与靶点活性位点的关键相互作用。显著稳定性提升,苗头化合物D6的抗DPP-IV降解能力提高40倍,血浆半衰期超过200小时,支持每周一次给药。D6在胰岛素抵抗的HepG2细胞中恢复胰岛素信号通路,并在db/db小鼠模型中实现长效降糖效果,疗效与司美格鲁肽相当。通过双脂肪酸共轭策略,首次实现了针对细胞内“不可成药”靶点PTPN1/PTPN2的长效肽类抑制剂开发,为代谢疾病治疗提供了新思路,并展示了该技术在靶向细胞内蛋白药物设计中的广泛应用潜力。
该工作以研究论文ARTICLE的形式在线发表于药学顶刊Journal of Medicinal Chemistry,青岛科技大学化工学院张传亮副教授和硕士研究生董国振为论文共同第一作者,通讯作者为青岛科技大学化工学院张传亮副教授和生物工程学院顾玉超教授。论文链接:https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.5c00147.。
进展二:脂肪酸化修饰虽能延长BH3多肽半衰期,但会降低水溶性,影响药物递送。因此,亟需一种既能提升肽类稳定性和长效性,又能改善其溶解性的新策略。基于此团队开发了“糖脂双修饰”技术,通过同时引入脂肪酸化和糖基化修饰,优化BimBH3肽的理化性质和药理活性。在BimBH3肽的N端连接棕榈酸(C16)或十八烷二酸(C18),并在Lys²侧链引入糖基(如唾液酸、半乳糖醛酸),显著提升水溶性(L3的溶解度提高10倍)和蛋白酶稳定性(抗DPP-IV降解能力提升4.4倍)。优化后的化合物L3在保持PTPN1抑制活性的同时,血浆半衰期延长至11.4小时(较原型肽提高9.85倍),解决了传统脂肽溶解性与稳定性不可兼得的矛盾。L3在口服葡萄糖耐量试验(OGTT)中表现出与司美格鲁肽相当的降糖效果,且长效性可持续10小时,支持其作为每周一次给药的潜力。同时,L3在口服糖耐量试验中持续改善胰岛素敏感性,也为开发口服肽类药物奠定基础。通过糖基化与脂肪酸化的协同作用,首次实现了BimBH3肽类抑制剂在溶解性、稳定性及生物活性上的全面优化,为开发靶向PTPN1的口服和长效抗糖尿病药物提供了新思路。双修饰策略的普适性也为其他肽类药物的设计提供了重要参考。
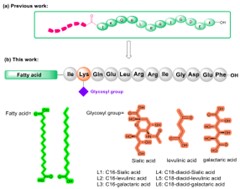
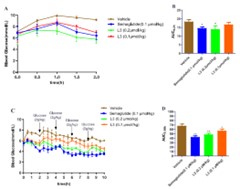
该工作以研究论文ARTICLE的形式在线发表于Bioconjugate Chemistry,青岛科技大学化工学院硕士研究生董国振为论文第一作者,通讯作者为青岛科技大学化工学院张传亮副教授。论文链接:https://pubs.acs.org/doi/10.1021/acs.bioconjchem.5c00057。
以上工作得到了研究得到了国家自然科学基金青年项目、山东省自然科学基金面上基金、青年项目及生态化工协同创新中心优秀人才基金的资助。
 化工学院微信
化工学院微信